Manual
Introduction
The leishMANIAdb contains experimental, computational and annotated information about proteins from Leishmania species. Currently the database focuses on five different species (L. brazliensis, L. donovani, L. infantum, L. major, L. mexicana).
Searching for specific proteins
Browsing the database
Users can also browse predefined sets of proteins. Currently proteins can be sorted based on species (L. brazliensis, L. donovani, L. infantum, L. major, L. mexicana) manual curation information, experimental information (secreted proteins, proteins with expression data, proteins with all kind of experimental data) and computationally predicted information (proteins that are expanded in Leishmania, transmembrane proteins, proteins with high disordered content, proteins with high-scoring linear motifs, proteins with PFAM domains).
Result summary page
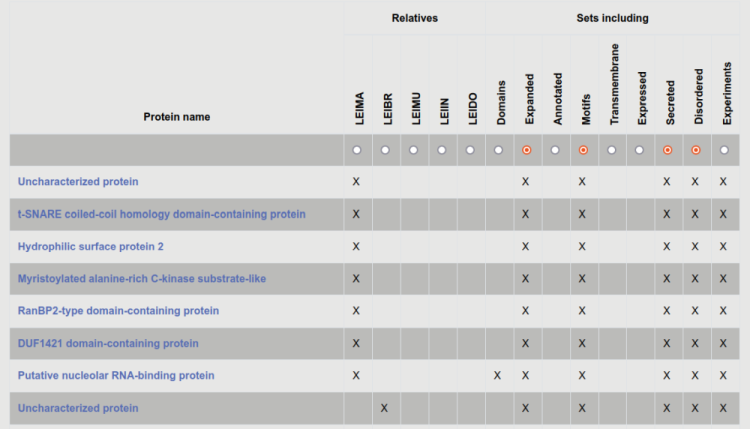
After searching for a protein or set of proteins, the user is directed to the result page, where basic information about the proteins are displayed. This includes species where homologous proteins can be found, and a quick review about what type of data is available for the protein (experimental and annotated). We also display predicted features, e.g. membrane proteins, disordered proteins can be selected. Users can further narrow results by selecting different features and annotation data on the top of the table.
For example, to search for secreted proteins with high-scoring motifs, disordered regions that are expanded in Leishmania, go to browse, select any of the aforementioned criteria, then on the result page click on the radio at the top of the table to select the remaining categories.
Download database
For each protein entry, annotations, structure predictions, the full multiple sequence alignment and the result of localization experiments can be downloaded at the bottom of the page. Users can also download the full database or predefined sets, using the Download menu in the top panel.
Entry page
1. Quick info
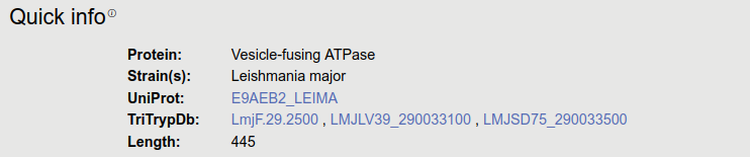
When selecting species, we faced that the quality of kinetoplastid proteomes vary. The two most widely used database for Trypanosoma protein research is probably UniProt and TriTrypDB. We found that several protein have different (mostly longer/shorter) sequences in the two databases. We decided to use UniProt as a reference, as many sequence and structure features, as well as crossreferences are more compatible with it.
We downloaded the Leishmania reference proteome from UniProt and TriTrypDB for five different species. In addition, we downloaded the following strains from these species: LbraziliensisMHOMBR75M2903, LbraziliensisMHOMBR75M2904, LbraziliensisMHOMBR75M2904_2019, LdonovaniBPK282A1, LdonovaniCL-SL, LdonovaniHU3, LdonovaniLV9, LinfantumJPCM5, LmajorFriedlin, LmajorFriedlin2021, LmajorLV39c5, LmajorSD75.1, LmexicanaMHOMGT2001U1103. If the protein sequence of UniProt and TriTrypDB is different: all information displayed is mapped to the UniProt version, however “*” marks TriTrypDB identifiers where users may find different sequences.
2. Annotations
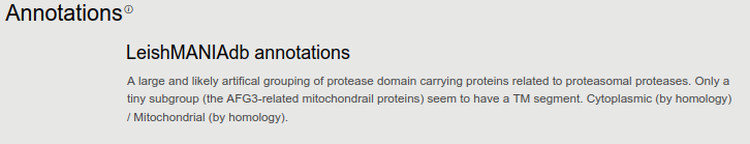
We found that direct information about virulence factors are very rare, only a handful of proteins were shown to interfere with host cell regulation. On the contrary, many times housekeeping and other functions are more easy to assign. Therefore annotations in this case may serve as a complementary approach - a sign of importance can be if no function is annotated, and experiments and predictions agree on a putative role in infection.
Our team annotated thousands of proteins with their putative function and localization. Since this type of annotation is often based on similarity search and transferring data, and only contains general information about a protein it is automatically included in all close relatives. We also collected data from literature from experiments on individual proteins. In this case the annotation is also displayed on homologs, but since the experiment was performed on a specific protein, on their homologs we display if the measurement was done on another protein.
Next to our own annotations, we also show the functional annotation by Jardim et al.
3. Localization
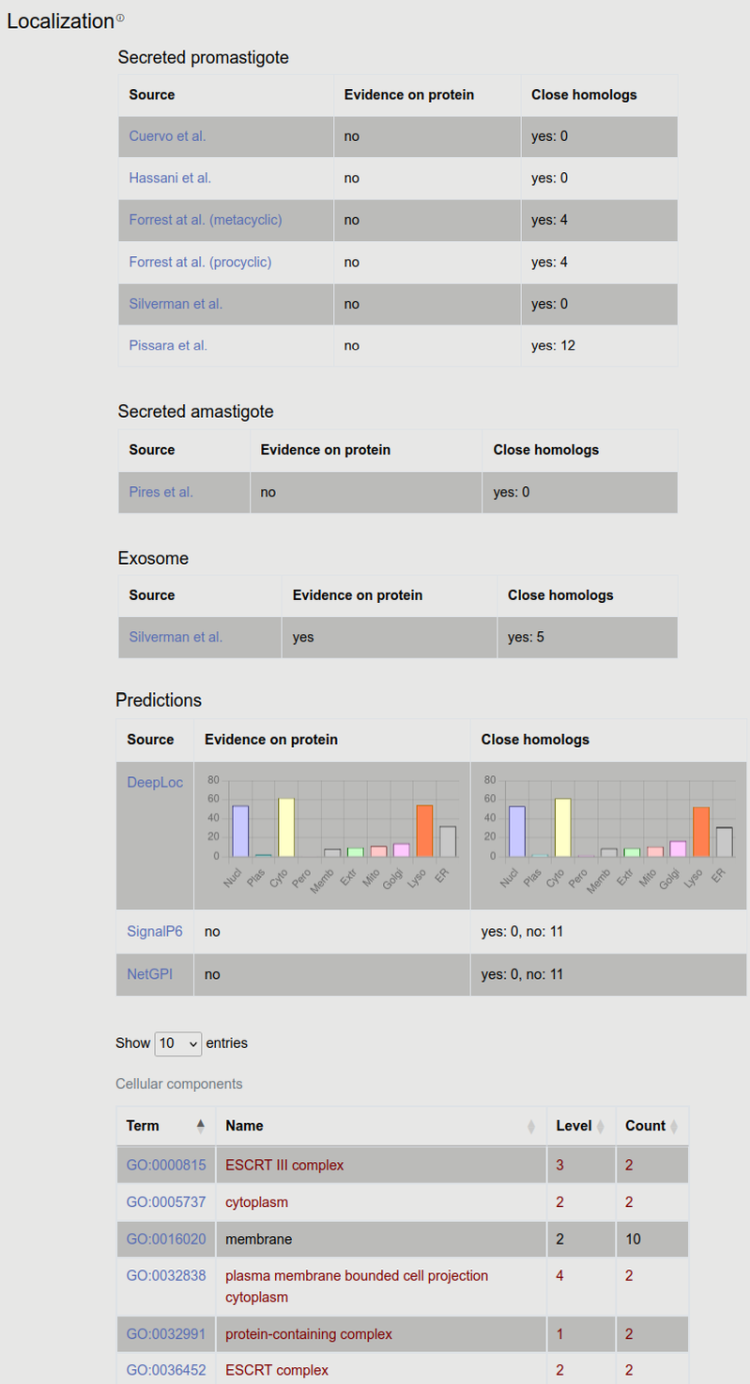
Dozens of high-throughput experiments were carried on in different Leishmania species, using different conditions and set-ups. Notably, even in promastigote experiments the authors used to expose the pathogen to a temperature and pH shift, therefore this information may be also valuable to understand the pathomechanism. Most of these experiments are expected to have some level of noise, and results shouldn’t be used without objective judgment. However if many experiments advocate the secretion (maybe supported by predictions), the protein may in fact act as a virulence factor. We also present results from homologous experiments and predictions - since the reliability is often lower (in both case), aggregating results may help to judge localization.
Information about protein localization can be found here. First we list experimental information from papers that measure which proteins are secreted in promastigote ( Cuervo et al, Hassani et al, Forrest et al, Silverman et al, Pisarra et al), or amastigote ( Pires et al) stage. If the protein is released in exosomes or it is glycosylated it is also displayed here. Next, prediction information is shown from DeepLoc (all kinds of localization), SignalP (signal peptide prediction) and NetGPI (GPI-anchor prediction). We also display all this data for homologous proteins (see later in Expansion) too, including experimental and prediction data.
We also show GeneOntology annotations for cellular compartments. In this case the specificity of the term (how deep it is on the tree) is shown in the “level” column. GO annotations are collected for all homologous proteins too, and the number of occurrences of each term is displayed. Red color highlights terms that are associated with the entry protein.
4. Abundance
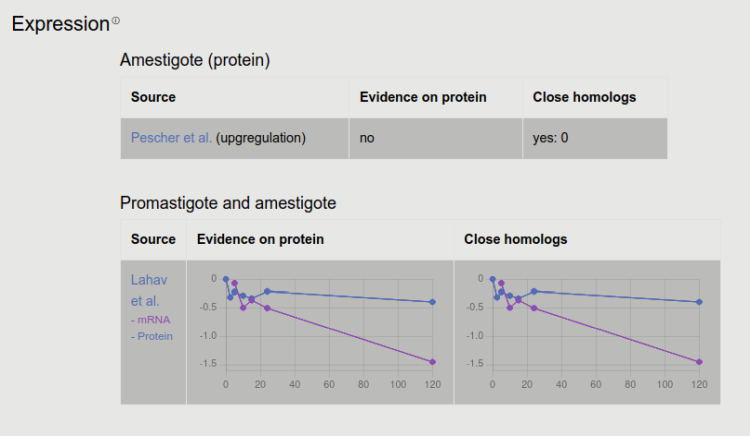
Two experimental datasets were incorporated. While Pescher and coworkers collected abundant proteins, Lahav et al measured mRNA and protein levels for a longer period at different times. Information from homologous proteins are also displayed here.
5. Expansion
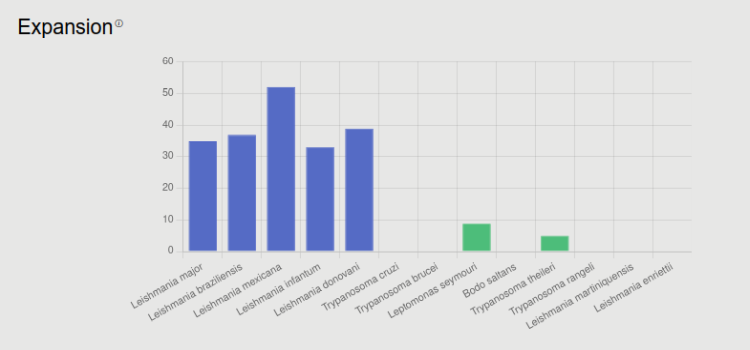
Leishmania species (and its close relative, T. cruzi) are intracellular pathogens, in contrast to several other kinetoplastids. Although such categorization is never binary, there is a definite distinction between parasites that grow and reproduce inside the host cell and other organisms. Expansion is expected to be higher when the protein function is important and unique for the pathogen, often resulting in gene duplication(s).
The number of homologous proteins from selected kinetoplastid species are displayed here. To find homologous, we performed BLAST search against SwissProt and the following reference proteomes:
- Bodo saltans
- Leishmania braziliensis
- Leishmania donovani
- Leishmania infantum
- Leishmania major
- Leishmania mexicana
- Leptomonas seymouri
- Trypansoma brucei
- Trypansoma cruzi
- Trypansoma rangeli
- Trypanosoma theileri
For each protein, we selected hits with the following thresholds: e-value: 10^-5; sequence identity>20%; coverage>50%. However, if we find a non-kinetoplastid hit, all other hits with lower sequence identities are omitted.
6. Sequence features
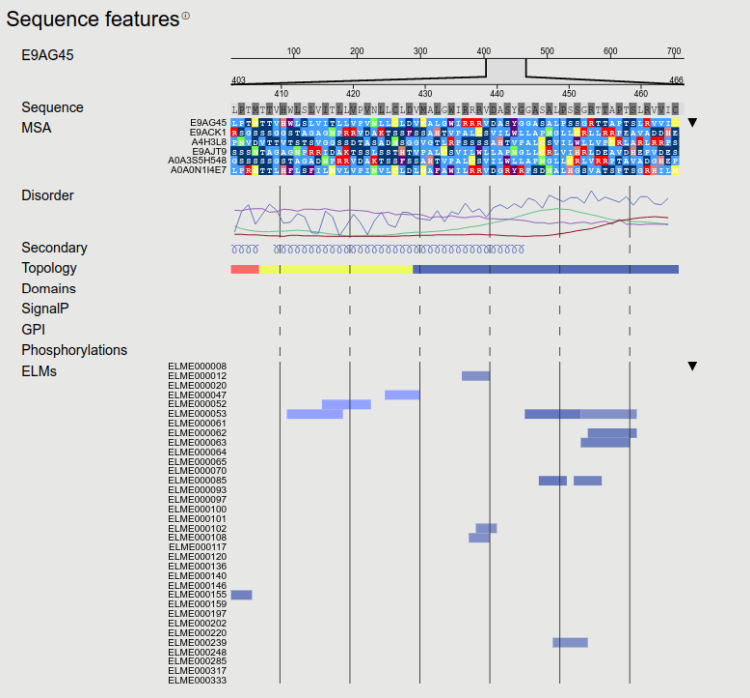
First the alignment of homologous proteins are displayed. The list of proteins included in the alignment is identical to the ones shown in the Expansion section. Since other features (see below) are displayed for the protein (without possible gaps originated from the alignment), here only the "gapless" alignment is shown (columns with gaps in the query proteins were removed). The full alignment file (that includes additional Leishmania strains) can be downloaded from bottom of the page.
Predicted sequence features are shown here. Transmembrane topology was predicted using CCTOP, however to avoid sporadic erroneous predictions, after an initial prediction we performed a constrained iteration too - where the topology of homologous proteins were used as a constraint. Using this approach closely related proteins will always have similar topology. Disordered regions were predicted by IUPred3 and using the AlphaFold models (if they are available). In the latter case we included pLDDT values and relative surface accessibility, as they were shown to correlate with protein disorder. For transmembrane proteins we also used MemDis, however in this case the prediction was tailored: the PSSM was calculated using kinetoplastid species, while accessibility and secondary structure information was taken from the AlphaFold models. From AlphaFold we also display secondary structures (using DSSP). Next PFAM domains are shown with crossreference to InterPro. Experiments performed by Tsigankov et al on stage dependent phosphorylation are displayed below domains. We also scanned proteins for Short Linear Motifs (SLiMs) using the regular expressions stored in the Eukaryotic Linear Motif resource. Scanning SLiMs alone will mostly yield false positive hits, so we developed a scoring system that takes into account the following information:
Residue level:
- conservation
- disordered/accessible regions
- localization
Protein level:
- expansion
- secretion
- expression
- homology to non-kinetoplastid proteins
We colored each SLiMs based on the score calculated. For details of the scoring see the following document.
Notably, we are aware the PFAM is currently not continued, however as the ELM resource is heavily dependent on PFAM (as the domain the SLiM binds to), we decided to keep it for now.
Users can use ctrl+mouse scroll to zoom into the sequence and highlight features. Arrows open the full panel for alignments and linear motifs.
7. Structure:
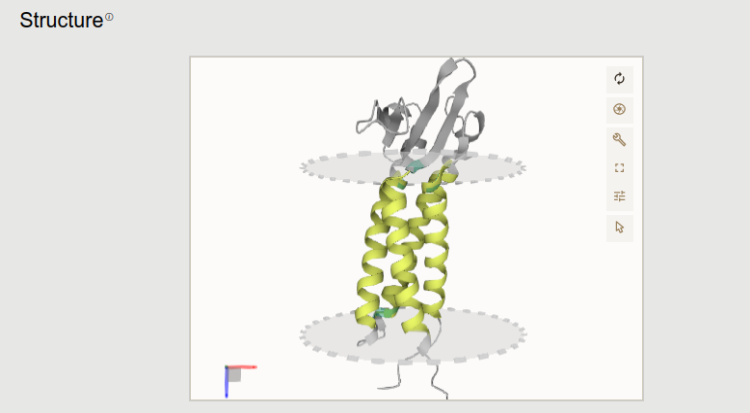
3D predicted structure from AlphaFold Protein Structure Database is visible here (note that it is available for around ~92% of the deposited Leishmania sequences). In case if it is a transmembrane protein, the lipid bilayer is also visible from TMAlphaFold database. Experimental structures from PDB is also displayed here.
8. Function
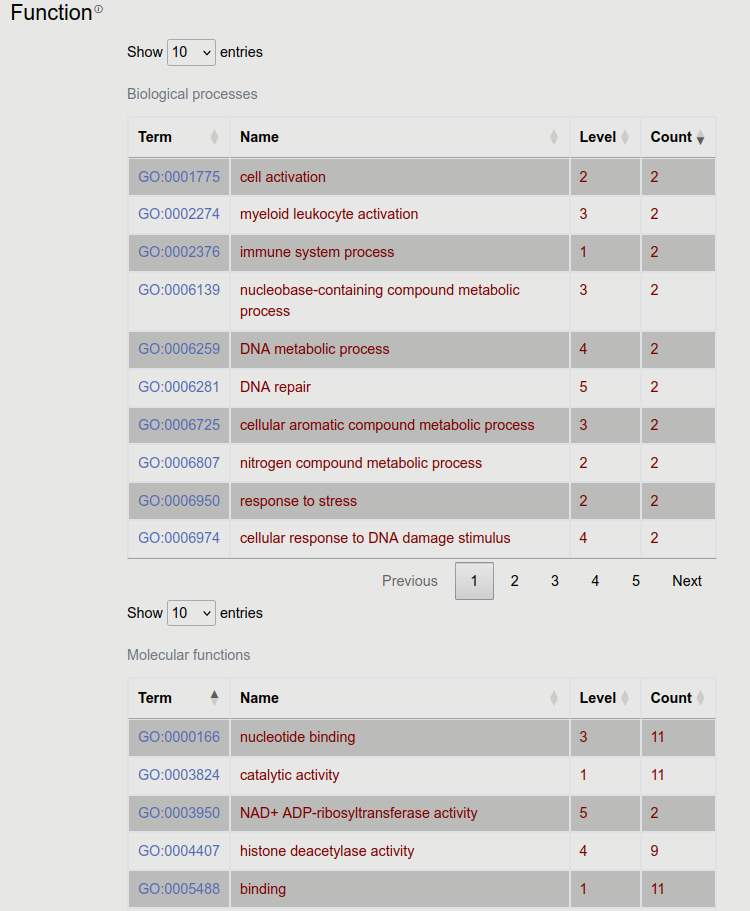
GeneOntology annotations for molecular function and biological process are visible here, in a similar manner as for cellular compartments (for more info see Localization).
9. Putative motif mimicry
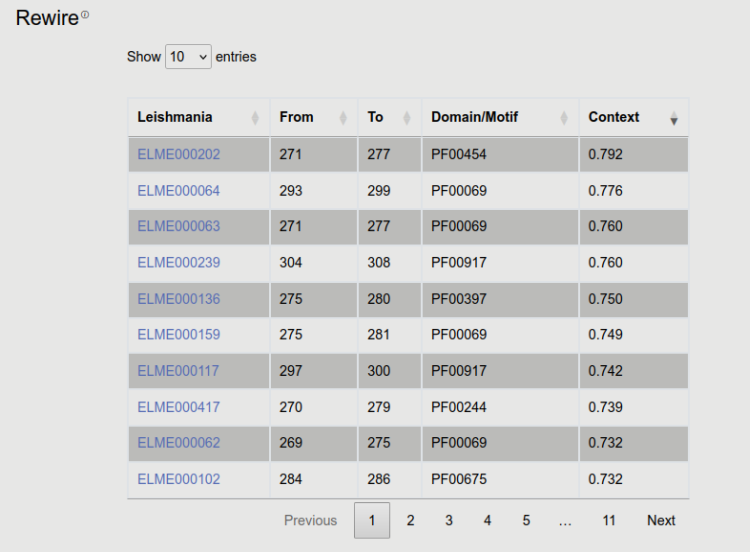
In this table all detected SLiMs are shown, together with start and end position, the corresponding PFAM domain and the score (see Sequence features for details). (Note that taxonomy filtering was also applied, therefore SLiMs not compatible with vertebrate host are excluded). For details of the scoring see the following document.
10. Homologs
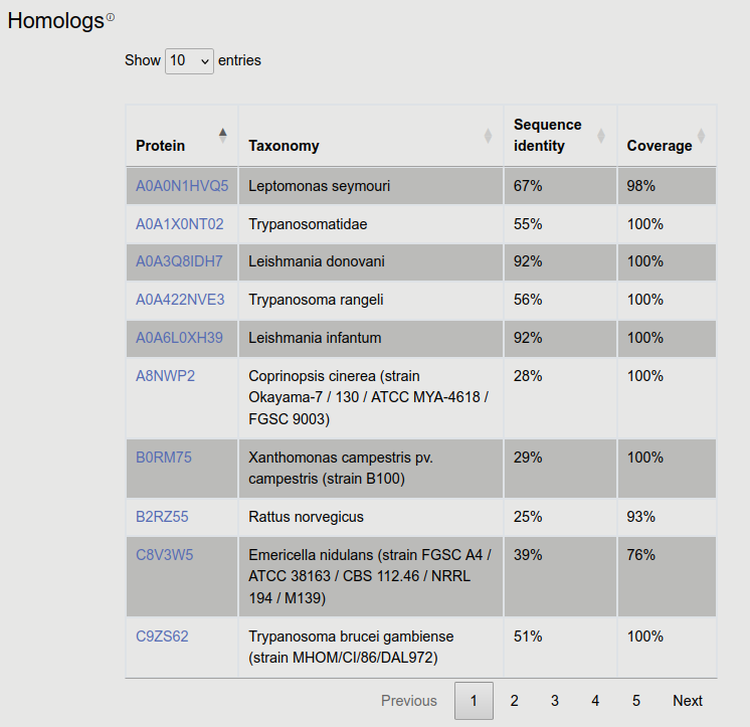
In this table we list each results from BLAST search agains SwissProt and kinetoplastid relatives, using the following thresholds: e-value: 10-5; sequence identity>20%; coverage>50%.
Panels are only visible if they contain data.